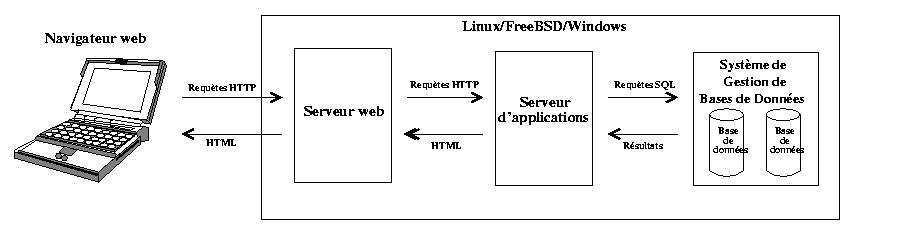
Introduction ŗ Perl et premiers pas
Perl possŤde plusieurs avantages : tout d'abord, il est installť sur la quasi-totalitť des environnements Unix. Ensuite, les scripts ne nťcessitent pas de compilation, et sont donc parfaitement portables d'un systŤme ŗ l'autre (Windows vers Linux par exemple). Perl implťmente ťgalement de nombreux types de donnťes trŤs utiles comme les tables de hachage (tableaux associatifs), qui ťvitent au programmeurs de rťinventer la roue. Il supporte les expressions rťguliŤres, ce qui lui permet d'effectuer des traitements compliquťs sur les chaines de caractŤres (recherche et remplacement de texte par exemple). Perl possŤde maintenant un nombre incalculable de modules, tous disponibles dans le doamine public, et couvre ainsi tous les domaines de l'informatique. Il est facile de dťbuter la programmation avec Perl.
Ouvrir avec Emacs un fichier nommť hello.pl, puis taper la ligne suivante, sauver le fichier puis sortir d'Emacs.
[olly@mplpc16 bioinfo]$ emacs hello.pl&
print "Hello!\n";
[olly@mplpc16 bioinfo]$ perl hello.pl
Maintenant, ouvrez le fichier avec emacs et ajoutez-y la ligne suivante :
#!/usr/bin/perl
$str = "Hello";
print "$str tout le monde!\n";
Ensuite, rendez le fichier ťxťcutable
[olly@mplpc16 bioinfo]$ chmod +x hello.pl
Vťrifions que le fchier est bien executable (il contient trois "x")
[olly@mplpc16 dtgen]$ ll hello.pl
-rwxr-xr-x 1 olly olly 18 Jun 5 23:19 hello.pl*
Et maitenant, on ťxťcute ce script!
[olly@mplpc16 dtgen]$ hello.pl
concepts fondamentaux de la syntaxe Perl
La syntaxe du Perl est relativement proche de celle du C (pour ceux qui connaissent ce langage). Ainsi, Perl est sensible ŗ la casse (print est diffťrent de PRINT par exemple). En Perl, chaque ligne est terminťe par un point-virgule. Tous ce qui suit le caractŤre # est considťre comme des commentaires.
Il existe trois types de variables en Perl : les scalaires, les listes et les hachages. Une variable de type scalaire dťbute par le signe $ (par exemple $count), une variable de type liste dťbute par le signe @ (par exemple @tableau), tandis qu'une variable de type hachage dťbute par le signe % (par exemple %hash).
Une variable de type scalaire peut contenir un nombre (entier ou rťel) ou bien une chaÓne de caractŤres. Par exemple :
$num = 12;
$taille = 3.3;
$str = "salut";
Perl rťalise l'interpolation de variable scalaire dans les chaÓnes de caractŤre. Par exemple le script suivant affiche le message "je mesure 1.87 m" :
$taille = 1.87;
print "je mesure $taille m";
Il n'y a pas de typage en Perl, cela signifie qu'une variable prend le type de ce qui lui est affectťe. Par exemple il est possible de faire les opťrations suivantes :
$var = 3
$var = "salut";
print $var;
ce qui affichera "salut" et non pas 3.
Comme dans de
construction de rťferences
AprŤs avoir passť en revue les constructions de bases, : Perl possŤde ťgalement comme particularitť de dťfinir une sťrie variables spťciales : $_
que veut dire "my"
syntaxe gťnťrale
Une liste (ou un tableau) est une variable contenant elle-mÍme une sťrie d'objets (variables, listes, etc.). Chaque objet (ou ťlťment) d'un tableau est repťrť par sa position au sein du tableau. Par exemple, le premier ťlťment d'un tableau est ŗ la position 0. Le cinquiŤme ťlťment est ŗ la position 4.
@tableau = (12, "abc", 56, "rtt");
print $tableau[0]; # affiche 12
print $tableau[3]; # affiche rtt
Il est possible avec la fonction map d'effectuer une ou plusieurs opťrations sur chacun des ťlťments d' un tableau
@tab2 = map {$_ * 2} @tab;
ou
@tab2 = map(function, @tab);
On peut ťgalement parcourir les ťlťments d'un tableau grace ŗ l'instruction foreach :
foreach $item (@tableau) {
print $item;
}
Les tableaux associatifs
De mÍme qu'un tableau, un tableau associatif associe une clť ŗ un ťlťment. En revanche la clť n'est pas restreinte ŗ un chifrre, il peut d'agir d'une chaine de caractŤres. Pour parcourir un tableau associatif nommť
%asso, on procŤdera de la maniŤre suivante :
foreach $item (%asso) {
# instructions ...
print $item;
}
Il existe une notion de contexte en Perl, c'est ŗ dire qu'il n'est pas toujours utile de spťcifier explicitement les paramŤtres d'une fonction
d'abord les regexp, puis les fichiers
Par exemple, la ligne suivante affichera la sťquence contenue dans $seq si celle-ci contient le motif TTATA :
print $seq if ($seq =~ /TTATA/)
L'interactivitť avec l'utilisateur est nťcťssaire pour bien des programmes conviviaux. Nťanmoins elle
Il y a deux moyens d'interagir avec des donnťes externes. La premiŤre consiste ŗ ouvrir et crťer des fichiers externes, la seconde ŗ utiliser les entrťes et sorties standard.
Perl gŤre de maniŤre simple les entrťes sorties standards...
trŤs utile pour rťaliser des traitements rapides sur des fichiers texte. Par exemple, supposons que l'on souhaite extraire les accession numbers du fichier suivant, nommť nucleo.seq :
>X14543
AATTTATGCGCTATATATATTTTG
>X14544
AATTTATGCGCTATGTATATTTTG
>X14545
AATTTATGCGCGATATATATTTTG
Le script numacc.pl permettant de rťaliser cette tache est le suivant :
while (<STDIN>) {
# affiche l'accession number si la ligne commence par >
print $1 if (/^>(.*)$/);
}
L'usage de ce programme est le suivant :
cat nucleo.seq | numacc.pl
ťcrire une nouvelle routine :
sub maRoutine {
my ($arg1, $arg2) = @_;
print "argument passťs : $arg1 et $arg2";
return $arg1+ $arg2;
}
parser les options de la ligne de commande avec le module Getopt::Long. Supposons que l'on souhaite demander ŗ l'utilisateur le type de sortie qu'il souhaite, et ce au travers de la ligne de commande :
script.pl --output=html
use Getopt::Long;
# definit le style de sortie, html par dťfaut
my $output = '';
GetOptions ('output=s' => \$output);
$output = "html" if ($output eq '');
if ($output eq "html") {
print "<b>Hello !</b>";
} else {
print "Hello !";
}
Par exemple, essayons d'extraire des sťquences les portions comprises entre les motifs TTA et TTC
Pour extraire les 10 premiers acides aminťes de chaque sťquence, on utilisera la fonction substr. Par exemple : substr($seq, 0, 10)
Les expressions rťguliŤres
Redirection des E/S
Dťfinition et utilisation des descripteurs de fichiers
APPLICATION DE MOTIFS ET OP…RATEURS
Expressions rťguliŤres Perl
Extraction d'informations textuelles importantes
Utilisation d'expressions rťguliŤres UNIX
Modification des donnťes avec des substitutions
Dťcodage des extensions "egimosx"
$ fin de ligne
? 0 ou 1 fois
^ dťbut de ligne
Installer un module Perl
Parser la sortie de Blast grŗce ŗ Perl
Si le module CPAN.pm est installť sur votre machine, il est possible d'installer de nouveaux modules de maniŤre quasi-automatique. Par exemple, pour installer le module SOAP::Lite, il suffit de taper la commande suivante (en tant que super-utilisateur):
perl -MCPAN -e 'install SOAP::Lite'
Notons que l'interface CPAN permet de chercher des modules Perl parmi ceux installables. Ainsi, pour chercher tous les modules contenant la chaÓne
perl -MCPAN -e 'shell'
i /Bio/
Autrement, il est nťcessaire de tťlťcharger l'archive en .tar.gz, de la decompresser, et de taper les commandes suivantes dans le rťpertoire crťť :
perl Makefile.PL
make
make test
su
make install
Ressources:
tutorial Perl en franÁais chez ftls.org http://www.ftls.org/fr/initiation/perl/
Un excellent Learning Perl O'Reilly
Perl permet de crťer et d'accťder ŗ la librairie GNU Database Manager. Il s'agit d'une librairie installťe sur tous les systŤmes Linux, dont les routines permettent de gťrer des fichiers contenant des paires clť/donnťes. On l'utilise couramment pour stocker des donnťes et pour pouvoir les rťcupťrer rapidement. Utile par exemple pour construire un petit moteur de recherche.
GNU dbm is a library of routines that manages data files that contain key/data pairs. The
access provided is that of storing, retrieval, and deletion by key and a non-sorted traver≠
sal of all keys. A process is allowed to use multiple data files at the same time.
Ecrire une base de donnťes:
use GDBM_File;
tie(my(%indexdb),'GDBM_File',"index.db", &GDBM_WRCREAT, 0644);
$indexdb{"bioinformatique"} = "file.html";
untie(%indexdb);
Lire une base de donnťes:
use GDBM_File;
tie(my(%indexdb),'GDBM_File',"index.db", &GDBM_WRCREAT, 0644);
$indexdb{"bioinformatique"} = "file.html";
untie(%indexdb);
La fonction
sort permet de trier un tableau, c'est ŗ dire de ranger les ťlťments de ce tableau dans un ordre spťcifiť. Par
exemple, supposons que l'on souhaite trier le fichier suivant, nommť specif.txt par ordre lexicographique :
Phenyl[1-(1-N-succinylamino)pentyl] phosphonate
Neuraminidase [influenza virus]; residues: 82-468
Lysozyme [hen egg white](HEL) EC:3.2.1.17 D18A
Cyclopentadienyl ferrocene (exo Diels-Alder reaction inhibitor)
Human rhinovirus (serotype 2) VP2 (viral capsid protein) peptide; residues: 156-170
Transition State Analogue (Bicyclo[2.2.2]octene derivative)
4-hydroxy-3-nitrophenyl acetate
4-hydroxy-5-iodo-3-nitrophenyl acetate
Aspartame
HIV-1 gp120; residues: 308-332
Autoantigen IgG4 Fc Rea
HIV-1 p24 (capsid protein); residues: 1-151
Thromboplastin (synonym: tissue factor, TF, coagulation factor III) extracellular domain
HIV-1 V3 loop constrained peptide analogue (Aib142)
5-(para-nitrophenyl phosphonate)-pentanoic acid
Cytochrome c oxidase [Paracoccus denitrificans] EC:1.9.3.1
2,2,6,6-tetramethyl-1-piperidinyloxy-dinitrophenyl
HIV-1 p24 epitope-homologous peptide
Lysozyme [bobwhite quail egg white] EC:3.2.1.17
Lysozyme [hen egg white]
Rnase A
Lysozyme [hen egg white] (HEL) EC:3.2.1.17
Traseolide [musk odorant fragrance] (hydrophobic hapten)
HIV-1 reverse transcriptase (HIV-1 RT) EC:2.7.7.49 M184I/double-stranded DNA (ds) complex
Le tri de ce fichier se fait en utilisant le programme Perl suivant :
#!/usr/bin/perl -w
@fichier = <STDIN>;
@result = sort @fichier;
print STDOUT @result;
Ligne de commande :
sort.pl < specif.txt > specif_triees.txt
Par dťfaut, le tri est effectuť par ordre lexicographique, mais
sort peut faire appel ŗ une fonction de comparaison externe. Par exemple, pour trier les lignes de ce fichier par ordre de repX croissant :
6 50
rep1 CTGACATTGGAAAGTACTTTATACGACAAGACAAACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
rep5 CTGACACCGGAAAGTACTTTATACGTCAAGACAAACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
rep2 CTGACATTGGAAAGTACTTTATACGTCGAGACGGACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
rep6 CTGACACCGGGGAGTACTTTATACGACAAGACAAACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
rep4 CTGACATTGGAAAGTACTTTATACGACAAGACAAACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
rep3 CTGACATTGGAAAGTACTTTATACGACAAGACAAACTGAAGGTGCAAGTCCTTTTTGTCATACTATTCAT
On utilisera le programme suivant :
#!/usr/bin/perl
#recupŤre la premiŤre ligne
$ln = <STDIN>;
#recupŤre les autres lignes
@lines = <STDIN>;
#sortie
print $ln;
@sorted_lines = sort sortlines @lines;
foreach $line (@sorted_lines) {
print "$line";
}
sub sortlines {
$a =~ /^rep(\d+)/;
$a1 = $1;
$b =~ /^rep(\d+)/;
$b1 = $1;
return $a1 <=> $b1;
}
Applications en bioinformatique
Mettre de l'information au format souhaitť
exemple : format bizarre -> format fasta
Rťcupťrer l'information ŗ partir de fichiers de sorties de programmes externes
Construction automatique de pages web
Le programme le plus simple possible est le suivant :
print "Bonjour je m'appelle Olivier\n";
Tout comme il est possible d'ťcrire du texte ŗ l'ťcran, il est ťgalement possible d'ťcrire du HTML :
print "<html><body>\n";
print "font color=\"red\">Bonjour je m'appelle Olivier</font>\n";
print "</body></html>\n";
Redirigeons la sortie de ce script vers un fichier nommť page.html (avec la commande print.pl > page.html), puis visualisons ce dernier avec netscape.
Notons que Perl permet de sťparer certaines donnťes du code HTML proprement dit, grace a l'usage de variables. Par exemple :
$couleur = "red";
$texte = "Bonjour je m'appelle Olivier";
print "<html><body>\n";
print "font color=\"$couleur\">$texte</font>\n";
print "</body></html>\n";
BioPerl correspond ŗ une sťrie de modules destinťs ŗ automatiser certaines taches communes en bioinformatique. Par exemple, il permet de lire des fichiers FASTA, de lancer des BLAST et de rťcupťrer des sťquences ŗ partir des bases de donnťes online.
L'installation de BioPerl peut se faire de la maniŤre suivante, via le module CPAN.pm :
[root@MPLPC18 olly]# perl -MCPAN -e shell
cpan shell -- CPAN exploration and modules installation (v1.52)
ReadLine support enabled
cpan> install "B/BI/BIRNEY/bioperl-0.7.1.tar.gz"
Un tutorial complet sur BioPerl est disponible ŗ l'adresse http://bio.perl.org/Core/POD/bptutorial.html
Rťcupťrer des sťquences ŗ partir de fichiers FASTA, EMBL, etc.
Exemple : lire et afficher les sťquences d'n fichier FASTA :
use Bio::SeqIO;
$seqio = Bio::SeqIO->new ( '-format' => 'Fasta' , -file => $ARGV[0]);
while $seq = $seqio->next_seq;
Autre exemple : lire et afficher les sťquences d'n fichierau format EMBL :
use Bio::SeqIO;
$seqio = Bio::SeqIO->new ( '-format' => 'embl' , -file => '../new_seq.dat');
while ($seqobj = $seqio->next_seq()) {
print ">" . $seqobj->accession_number . "\n";
print $seqobj->seq . "\n";
}
Meme chose, mais avec stockage des sequences dans une base GDBM :
use Bio::SeqIO;
use GDBM_File;
tie(my(%indexdb),'GDBM_File',"sequences.db", &GDBM_WRCREAT, 0644);
$seqio = Bio::SeqIO->new ( '-format' => 'embl' , -file => '../new_seq.dat');
$cnt = 0;
while (($seqobj = $seqio->next_seq()) && ($cnt++ < 100)) {
print ">" . $seqobj->accession_number . "\n";
print $seqobj->seq . "\n";
$indexdb{$seqobj->accession_number} = $seqobj->seq;
}
untie(%indexdb);
Lecture des infos stockťes :
use GDBM_File;
tie(my(%indexdb),'GDBM_File',"sequences.db", &GDBM_WRCREAT, 0644);
while (($k, $v) = each %indexdb) {
print ">" . $k . "\n";
print $v . "\n";
}
untie(%indexdb);
Rťcuperer des sťquences dans GenBank
use Bio::DB::GenBank;
$gb = new Bio::DB::GenBank;
$seq = $gb->get_Seq_by_acc($ARGV[0]);
print $seq->seq;
Utiliser le module d'alignement multiple (SW)
#!/usr/bin/perl
use Bio::Tools::pSW;
use Bio::Seq;
$factory = new Bio::Tools::pSW( '-matrix' => 'blosum62.bla',
'-gap' => 12,
'-ext' => 2, );
$seq1 = Bio::Seq->new( -seq => 'WLGQRNLVSSTGGNLLNVWLKDW');
$seq2 = Bio::Seq->new( -seq => 'WMGNRNVVNLLNVWFRDW');
$factory->align_and_show($seq1, $seq2, STDOUT);
$aln = $factory->pairwise_alignment($seq1, $seq2);
Ce qui devrait donner la sortie suivante :
seq1 1 WLGQRNLVSSTGGNLLNVWLKDW
W+G RN+V NLLNVW +DW
seq2 1 WMGNRNVV-----NLLNVWFRDW
Batir une interface graphique avec TK
#!/usr/bin/perl -w
use Tk;
use strict;
new MainWindow -> Button(-text => 'Press me!', -command => sub{print "Merci\n"})
-> pack;
MainLoop;
comp.lang.perl.tk FAQ
Ressources: